Reaction: Meta publishes ESMFold, a model that predicts the structure of hundreds of millions of proteins
Meta has applied language modelling to predict the structure of a large collection of proteins. The model, called ESMFold, is being presented this week in the journal Science after being published on the bioRxiv preprint article server in December 2022. EMSFold is faster than similar models such as AlphaFold, developed by Google's DeepMind and EMBL's European Bioinformatics Institute. The sequences of more than 617 million proteins - of which more than a third are predicted with a high degree of confidence - are published in open access in the ESM Metagenomic Atlas.
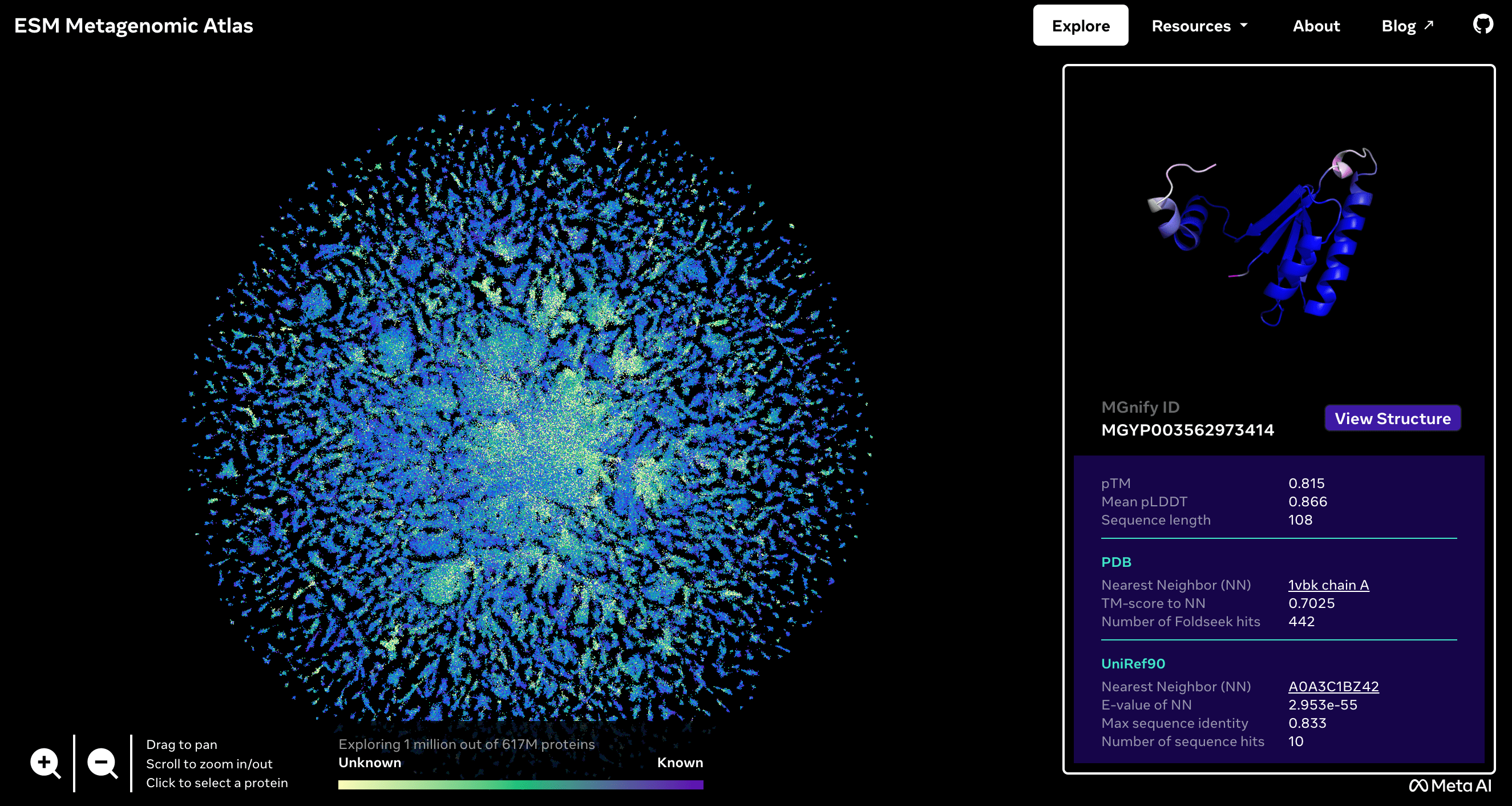
ESM Metagenomic Atlas.
Alfonso Valencia - Meta IA EN
Alfonso Valencia
ICREA professor and director of Life Sciences at the Barcelona National Supercomputing Centre (BSC).
Alexander Rives and colleagues present the results of the application of Large Language Models to predict protein structure. Although it is not the first work of this kind, it has produced a larger model size and a larger set of predicted protein structures.
Language models are statistical artefacts that exploit correlations between elements in large data sets to deduce the most likely chain of elements. The principle is the same as the already popular ChatGPT—in this case, applied to chains of amino acids (a 20-letter code) that make up proteins, rather than the characters of a human language.
The new model—it is the third version, nothing new in this sense—was trained using 138 million known protein sequences in order to predict the next amino acid in a sequence and, surprisingly, also to predict the corresponding three-dimensional structure. The surprise is only apparent, though: the logic of the succession of amino acids in known proteins is the result of an evolutionary process that has led them to have the specific structure with which they perform a particular function. This is the secret of these systems, their beauty is their conceptual simplicity. In short, trained with many proteins, the language model is able to predict the corresponding structure for a known chain. The process directly relates the DNA reading of a gene to the structure of the corresponding protein, closing the loop of information coding from DNA to proteins.
The quality of the results, validated with two sets suitable for this task (CAMEO and CASP14) is sufficiently convincing. The first difference with previous proposals based on deep neural networks (AlphaFold and RoseTTAFold) is that the new models are much easier to compute and much faster: between one and two orders of magnitude. As a demonstration of this capability, the study includes models for more than 617 million proteins. For this full set, the results are a little lower in quality than those obtained with previous methods, but they are comparable if we only take into account the structures predicted with high enough confidence (about 225 million).
The confidence in each prediction is a parameter derived from the fundamental model for the practical use of the results. It’s interesting that more than 10% of the predictions are for proteins that bear no resemblance to other known proteins. This is possible for language models that, unlike previous methods, do not require information from nearby sequences (sequence alignments). This means the new method can be applied directly to predicting the consequences of point mutation; this was beyond the scope of previous methods and has a direct impact on biomedical applications.
In addition, in their subsequent work, the authors have used this property to predict the structure of non-natural proteins, expanding the known universe beyond what the evolutionary process has explored. This is a new space for the design of proteins with new sequences and biochemical properties with applications in biotechnology and biomedicine.
Finally, for those of us who have been working in this field for the past 40 years, it is very surprising that large technology companies are investing all these efforts in a research topic that was considered a marginal, theoretical area. It is easy to think that this is a challenge between companies—Meta and Google/DeepMind. It is interesting that both have made their software and results openly available, something that is fairly unusual for these companies. Beyond the possible competition between the two companies, perhaps the real reason for the interest in this topic is much more technical: protein structure prediction seems to be a more suitable field for refining language modelling technologies than the generation of texts, which was their initial field of development. On the one hand, protein sequences are more abundant than texts, have more defined sizes and a higher degree of variability. On the other hand, proteins have a strong internal ‘meaning’—that is, a strong relationship between sequence and structure, a meaning or coherence that is much more diffuse in texts.
Zeming Lin et al.
- Research article
- Peer reviewed