Reacción: Meta publica ESMFold, un modelo que predice la estructura de cientos de millones de proteínas
La empresa Meta ha aplicado modelos de lenguaje para predecir la estructura de una gran colección de proteínas. El modelo, llamado ESMFold, se presenta esta semana en la revista Science tras ser publicado en el servidor de artículos preprint bioRxiv en diciembre de 2022. EMSFold es más rápido que otros modelos parecidos como AlphaFold, desarrollado por la empresa DeepMind de Google y el Instituto Europeo de Bioinformática del EMBL. Las secuencias de más de 617 millones de proteínas —de las cuales más de un tercio son predichas con un alto grado de confianza— se publican en acceso libre en el ESM Metagenomic Atlas.
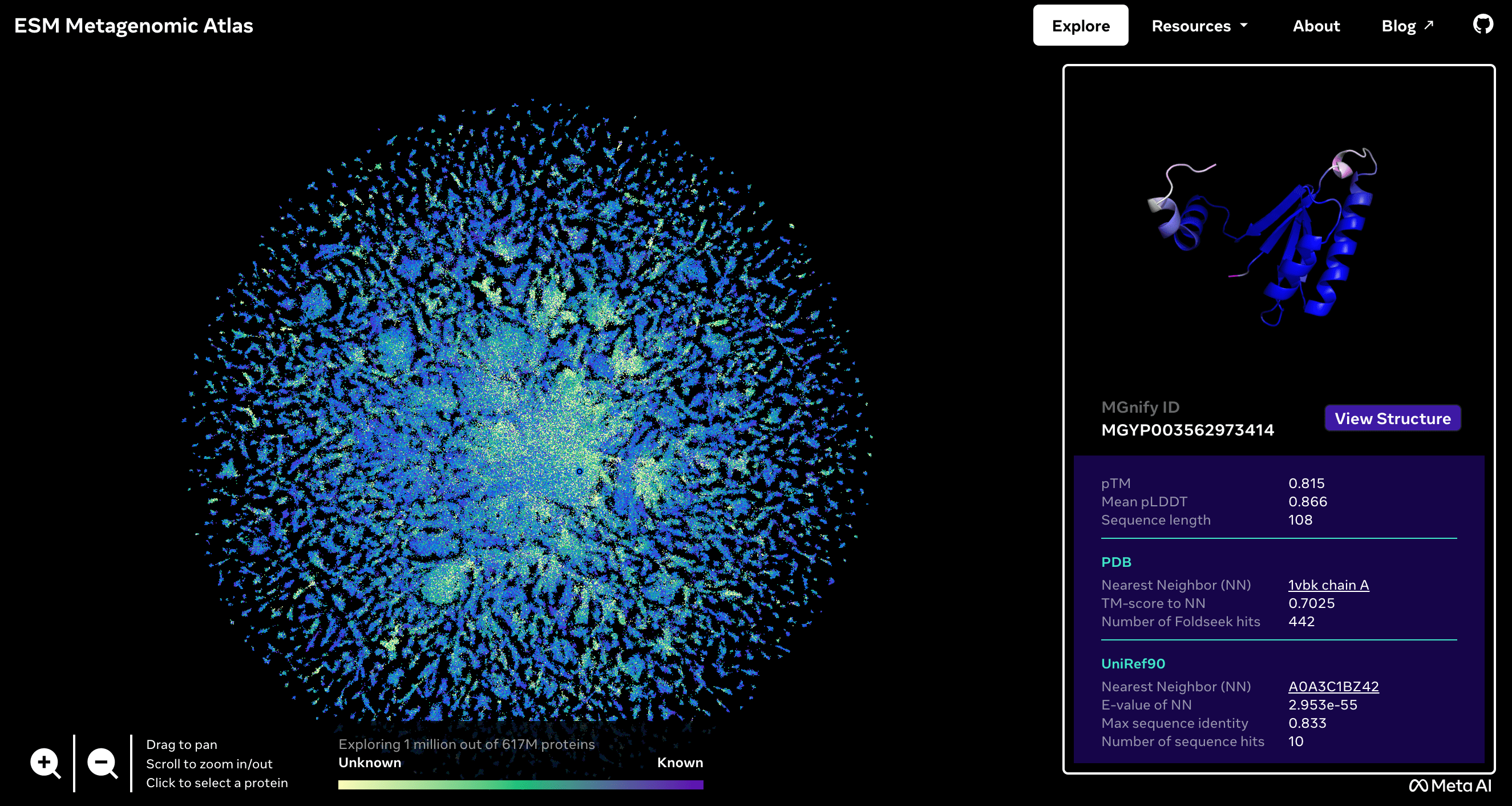
ESM Metagenomic Atlas.
Alfonso Valencia - Meta IA
Alfonso Valencia
Profesor ICREA y director de Ciencias de la Vida en el Centro Nacional de Supercomputación de Barcelona (BSC)
Alexander Rives y colaboradores presentan los resultados de la aplicación de modelos de lenguaje (Large Language Models) a la predicción de estructura de proteínas. Aunque no es el primer trabajo en esta línea, sí es el que ha producido un modelo de mayor tamaño y un conjunto mayor de estructuras de proteínas predichas.
Los modelos de lenguaje son artefactos estadísticos que explotan las correlaciones entre elementos de grandes conjuntos de datos para deducir la cadena más probable de elementos. El principio es el mismo del ya popular ChatGPT, en este caso, aplicado a cadenas de aminoácidos (un código de 20 letras) que forman las proteínas en vez de los caracteres de un lenguaje humano.
El nuevo modelo —es la tercera versión, nada nuevo en este sentido—, entrenado con unos 138 millones de secuencias de proteínas conocidas, aprende a predecir el siguiente aminoácido y de modo sorprendente también la estructura tridimensional correspondiente. La sorpresa es solo aparente, la lógica de la sucesión de los aminoácidos en las proteínas conocidas es el resultado del proceso evolutivo que las ha llevado a tener la estructura concreta con la que desarrollan una función determinada. Este es el secreto de estos sistemas, su belleza es su simplicidad conceptual. En resumen, entrenado con muchas proteínas, el modelo de lenguaje es capaz de producir la estructura correspondiente para una cadena conocida. El proceso relaciona directamente la lectura del ADN de un gen con la estructura de la proteína correspondiente, cerrando el circulo de codificación de la información del ADN a las proteínas.
La calidad de los resultados, validados con dos conjuntos adecuados para esta tarea (CAMEO y CASP14) es suficientemente convincente. La primera diferencia con las propuestas anteriores basadas en redes neuronales profundas (AlphaFold y RoseTTAFold) es que los nuevos modelos son mucho más fáciles de calcular y mucho más rápidos —entre uno y dos órdenes de magnitud—. Como demostración de esta capacidad, el estudio libera modelos para más de 617 millones de proteínas. En este conjunto completo los resultados son un tanto peores que los obtenidos con los métodos anteriores, aunque se vuelven comparables si se toman en cuenta solo las estructuras predichas con suficiente confianza (unos 225 millones).
La confianza en cada predicción es un parámetro derivado del modelo fundamental para la utilización práctica de los resultados. Es interesante que más del 10 % de las predicciones corresponden a proteínas que no tienen ningún parecido con otras conocidas, algo posible para los modelos de lenguaje que, a diferencia de los métodos anteriores, no requieren información de secuencias próximas (alineamientos de secuencias). Esto hace que la nueva metodología sea directamente aplicable a la predicción de las consecuencias de mutaciones puntuales, algo que estaba fuera del alcance de métodos anteriores y tiene un impacto directo en las aplicaciones en biomedicina.
Adicionalmente, en trabajos posteriores, los autores han utilizado esta propiedad para predecir la estructura de proteínas no naturales expandiendo el universo conocido más allá de lo que el proceso evolutivo ha explorado. Un espacio nuevo para el diseño de proteínas con nuevas secuencias y propiedades bioquímicas con aplicaciones en biotecnología y biomedicina.
Finalmente, para los que hemos trabajado en este campo durante los últimos 40 años es muy sorprendente que grandes compañías tecnológicas inviertan todos esos esfuerzos en un tema que se consideraba minoritario y teórico. Es fácil pensar que se trata de un reto entre compañías, Meta y Google/DeepMind. En este sentido es interesante que ambas compañías han hecho el software y los resultados disponibles abiertamente, algo no tan habitual en estas compañías. Más allá de las posibles razones de competencia entre compañías, quizás la razón real de su interés en este tema sea mucho más técnica; la predicción de estructura de proteínas parece ser el campo más adecuado para perfeccionar las tecnologías de modelos de lenguaje que la generación de textos donde se desarrollaron inicialmente. Por una parte, las secuencias de proteínas son más abundantes, tienen tamaños más definidos que los textos y presentan un grado de variabilidad mayor. Por otro lado, las proteínas tienen un fuerte ‘significado’ interno, esto es, una fuerte relación entre secuencia y estructura, un significado o coherencia que es mucho más difuso en los textos.
- Artículo de investigación
- Revisado por pares
Zeming Lin et al.
- Artículo de investigación
- Revisado por pares